

Leukemia driver mutations have been identified in clonal hematopoiesis (CH; Jaiswal et al, NEJM, 2014). This provides a window of opportunity to interrogate the downstream impact of driver mutations in the earliest stages of neoplasia, before the accumulation of additional drivers that lead to frank malignancy. However, CH mutated cells are morphologically and phenotypically similar to normal cells. Thus, previous characterization in primary human samples have largely focused on genetic identification of these clonal outgrowths.
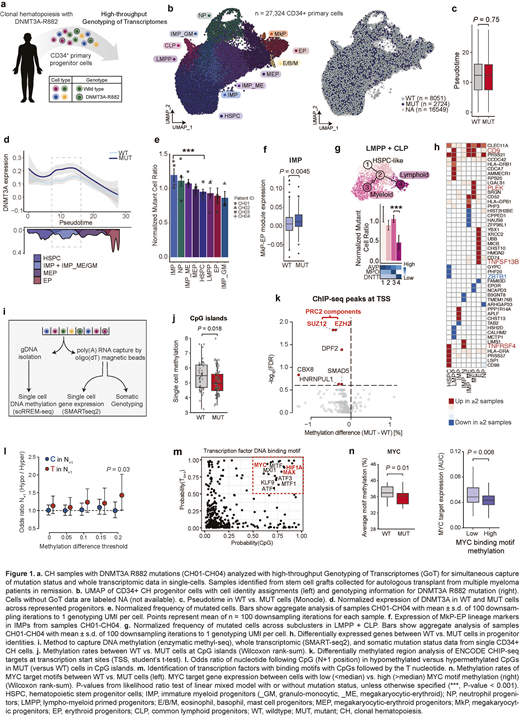
To overcome this limitation and define the downstream effects of CH mutations, we leveraged multi-omic single-cell sequencing to profile the mutation status and whole transcriptome for the same cells at high-throughput (GoT; Nam et al, Nature, 2019). To identify CH samples, we screened 136 stem cell grafts collected for autologous transplant from multiple myeloma patients in remission (Mouhieddine et al, Nat Comm, 2020). We identified four individuals with DNMT3AR882 mutations with VAF > 0.05, allowing the profiling of 27,324 CD34+ cells (Fig. 1a-b).
DNMT3AR882 mutations in these human CH samples did not result in a significant differentiation block in the hematopoietic stem cells, as assessed by pseudotime analysis (Fig. 1c). However, specific differentiation skews were apparent in multi-lineage progenitors, corresponding to the highest expression of DNMT3A in mutated immature myeloid progenitors (IMP, i.e. common myeloid progenitors/granulocyte-monocyte progenitors, Fig. 1d). We observed the expansion of mutated IMPs (Fig. 1e) that showed priming toward the megakaryocytic-erythroid (ME) lineage (Fig. 1f). DNMT3AR882 lympho-myeloid primed progenitors showed a myeloid bias and disfavored lymphoid development (Fig. 1g).
In these CD34+ progenitors, we identified dysregulated expressions of megakaryocytic lineage markers, such as CD9 and PLEK, consistent with the ME-bias (Fig. 1h). We also observed downregulation of lymphoid differentiation gene ZBTB1, corresponding to the myeloid over lymphoid skewing (Fig. 1h). Finally, we identified dysregulated expression of genes involved in TNF-alpha signaling (e.g. TNFRSF4, TNFSF13B), suggesting a pro-inflammatory state. To identify activated pathways, we performed a gene set enrichment analysis that revealed activation of MYC and IL-6 signaling (FDR <0.05, Hallmark). A focused analysis of regulatory networks identified enhanced activity of key transcription factors (TFs) involved in ME differentiation such as GATA1 and FLI1, as well as those involved in inflammation such as NFKB/REL (FDR <0.05). We also identified increased expression of polycomb repression complex 2 (PRC2) target genes (FDR <0.05).
To determine how aberrant DNA methylation may serve as a link between DNMT3A mutations and the observed transcriptional dysregulation, we performed single-cell multi-omics that simultaneously profiles the cells' methylome and transcriptome, linked with mutation status (Gaiti et al, Nature, 2019; Fig. 1i). By comparing the methylation rates in mutated vs. wildtype CD34+ cells within the same sample, we found that DNMT3AR882 result in selective hypomethylation that preferentially impacts CpG islands (Fig. 1j) and PRC2 targets (Fig. 1k), which may underlie the dysregulated expression of PRC2 targets in DNMT3AR882 progenitors. Notably, DNMT3AR882-induced hypomethylation was enriched in a specific sequence context in which the CpG is followed by a T nucleotide (Fig. 1l). This DNMT3AR882 motif was enriched in the DNA binding motifs of central hematopoietic TFs, including MYC/MAX (Fig. 1m), suggesting that DNMT3AR882-induced hypomethylation may serve as a mechanism to preferentially increase the activity of these TFs. Consistent with this hypothesis, joint single-cell methylome and transcriptome data revealed that the expression of MYC targets increased with hypomethylation of its binding motifs (Fig. 1n), providing a mechanism of enhanced MYC activity due to DNMT3AR882 mutations.
Altogether, we report the direct examination of the consequences of DNMT3AR882 mutations in primary CD34+ cells in CH. We discovered that DNMT3A is most highly expressed in mutated multi-lineage progenitors, promoting their expansion and biasing their downstream differentiation. This is accompanied by a disruption to differentiation through PRC2 and MYC target reactivation via selective hypomethylation.
Abdel-Wahab:Envisagenics Inc.: Current equity holder in private company; H3 Biomedicine Inc.: Consultancy, Research Funding; Janssen: Consultancy; Merck: Consultancy. Ghobrial:Takeda: Consultancy, Honoraria; Janssen: Consultancy, Honoraria; Bristol-Myers Squibb: Consultancy, Honoraria; Celgene: Consultancy, Honoraria; Amgen: Consultancy, Honoraria; Karyopharm Therapeutics: Consultancy, Honoraria; Cellectar: Honoraria; Adaptive Biotechnologies: Consultancy, Honoraria; Sanofi: Consultancy, Honoraria; Novartis: Consultancy; Noxxon Pharma: Consultancy; Genentech: Consultancy; GlaxoSmithKline: Consultancy; GNS Healthcare: Consultancy; AbbVie: Consultancy. Landau:Bristol Myers Squibb: Research Funding; Illumina: Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal